GLENN LOPATE, MD, ALAN PESTRONK, MD, MUHAMMAD AL-LOZI, MD,
TIMOTHY LYNCH, MD, JULAINE FLORENCE, DPT, TIMOTHY MILLER, MD,
TODD LEVINE, MD, TOM RAMPY, MD, BRENT BESON, MD, and IRENA RAMNEANTU, MD
ABSTRACT: Peripheral neuropathy is common in patients with Sjögren’s syndrome (SS), but its precise prevalence is unknown. Most prior studies were conducted at neurology or rheumatology specialty clinics and likely selected for a more severely affected population. We evaluated 22 SS patients and 10 controls for evidence of neuropathy in an outpatient setting at a regional meeting of the Sjögren’s Syndrome Foundation. We performed neurological examinations and nerve conduction studies (NCSs) and measured serum antinuclear antibody (ANA) and SS-A and SS-B antibody levels. Participants filled out a questionnaire pertaining to symptoms, diagnosis, and treatment. We found that signs and symptoms related to small axons were more common in patients with SS than in controls. Complaints of painful distal paresthesias in the feet were noted in 59% of patients but in only 10% of controls, and of abnormal sweating in 41% and 0%, respectively. Examination revealed decreased pinprick sensation in 64% of patients with SS, but in only 30% of controls. Overall, 45% of the patients but none of the controls were thought to have an isolated small-fiber neuropathy. Large-fiber dysfunction (as measured by testing vibration, deep tendon reflexes, and NCSs) was similar between the two groups. We conclude that small-fiber neuropathy is common in patients with SS.
Sjögren’s syndrome (SS) is a chronic inflammatory disease characterized by lymphocytic infiltration of the salivary, lacrimal, and other exocrine glands. Extraglandular involvement is common, including the peripheral nervous system, but the true prevalence of peripheral neuropathy is not known. Estimates vary from 10% to 60%, depending on the patient population, definition of neuropathy, and method of detection.1–3,5,8,10,11,13,15 Several different types of neuropathy have been described, with the most common being a distal sensorimotor polyneuropathy.1–3,5,8,10 –13,16 Other neuropathies include a sensory neuropathy/neuronopathy,1,5,7,10 –13,16 carpal tunnel syndrome,1,2,10,13 small-fiber neuropathy,6,9,16 mononeuritis multiplex,1,5,11,16 demyelinating neuropathy,11,13 motor neuropathy,1,13 autonomic neuropathy,5,11,16 and cranial neuropathy.1,5,10,11,13,15,16 Most previous studies on the type and prevalence of neuropathy have been conducted in university associated specialty clinics, where patients with SS are screened for the presence of neuropathy, or patients with SS and neurologic complaints are selected for further evaluation. This may cause a selection bias to include patients with more severe disease who seek out specialized care. We had the opportunity to examine and perform electrodiagnostic and laboratory studies in an outpatient setting on a prospective series of patients with SS. We report on the frequency and type of neuropathy in this population with comparison to a control group of similar age.
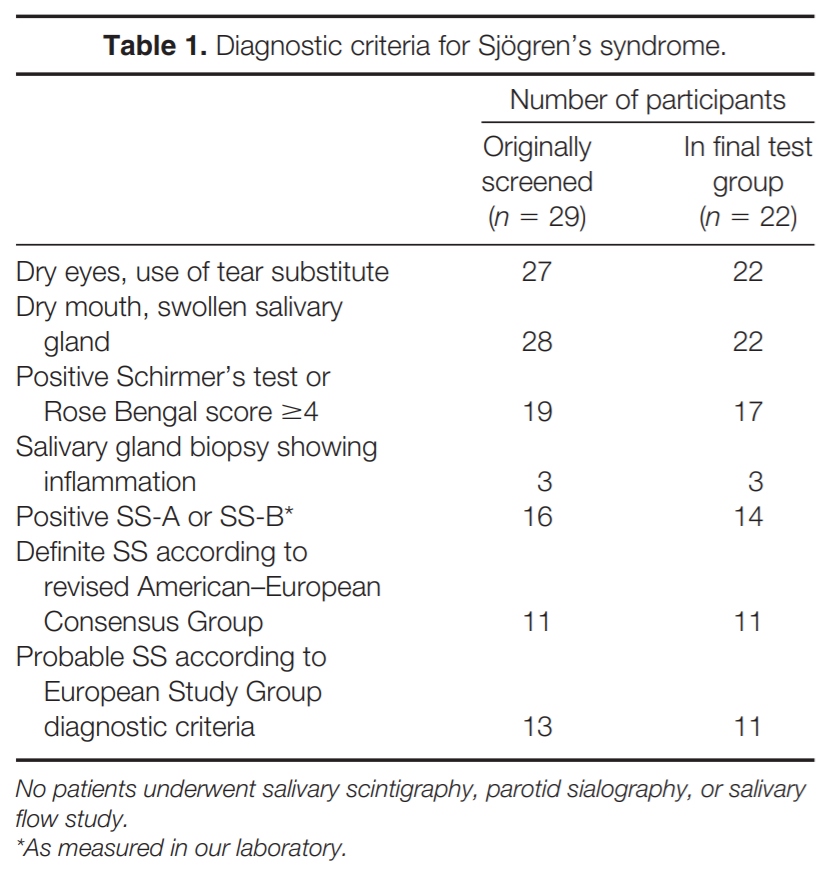
METHODS
Subjects. Twenty-nine patients and 11 spouses/significant-others attending a regional meeting of the Sjögren’s Syndrome Foundation in November 2003 completed our entire evaluation. The study was approved by our institutional review board and all participants provided informed consent. The diagnosis of SS was based on patient history in relation to criteria originally proposed by the European Study Group on Diagnostic Criteria for SS18 and modified by the American–European Consensus Study Group on Diagnostic Criteria for SS17 (see Questionnaire subsection). A total of 24 of 29 (83%) of the patients who were screened for a diagnosis of SS fulfilled the criteria (Table 1); 11 fulfilled the revised American– European Consensus Group criteria for the diagnosis of definite SS, and 13 patients had three positive items and fulfilled the European Study Group criteria for probable SS. Two patients had secondary SS and were excluded from further analysis, 1 with a diagnosis of rheumatoid arthritis (this patient did not meet criteria for SS) and 1 with systemic lupus erythematosus. One additional patient was also excluded from further comparisons because of an alternative cause for the neuropathy, non–insulin dependent diabetes mellitus. One control patient with non–insulin-dependent diabetes mellitus was also excluded from analysis. All comparisons detailed in the Results section are made between the final group of 22 patients with SS (11 definite SS, 11 probable SS) and the 10 controls (Table 1). Eight of the 22 patients with SS had positive serum antinuclear antibodies (ANA) detected in our laboratory. No patients or controls had antibodies in serum to sulfatide, myelin-associated glycoprotein, or GM1 ganglioside. None of the control patients had positive SS-A or SS-B and only 1 had positive ANA.
Neurologic Examination. A complete neurologic examination was performed by board-certified neurologists who were blinded to the diagnosis of SS and the results of electrodiagnostic testing. In addition, vibration was quantified using a Rydel–Seiffer tuning fork bilaterally at the toes, ankles, and hands by a different neurologist, who was also blinded to other testing. Based on previous studies,14 a value ≥8 (sum of both sides) was considered normal for the toes if age was ≤60 years and a value ≥7 (sum of both sides) if age was >60 years. A diagnosis of small-fiber neuropathy was made when a patient complained of distal painful paresthesias combined with abnormal pinprick testing. A diagnosis of large-fiber neuropathy was made when a patient complained of distal paresthesias or numbness and, on examination, had either decreased distal vibration by Rydel–Seiffer tuning fork and decreased or absent deep tendon reflexes (DTRs), or, on electrodiagnostic testing, showed low amplitudes of sural sensory nerve action potentials (SNAPs) or peroneal compound muscle action potentials (CMAPs).
Electrodiagnostic Studies. Nerve conduction studies (NCSs) were performed by a board-certified electrodiagnostic medicine consultant using standard techniques. Sural SNAPs and conduction velocities, and peroneal CMAPs, distal latencies, and foreleg conduction velocities were recorded from all participants. Additional studies of median and ulnar sensory and of median, ulnar, and tibial motor nerves were done on an individual basis as deemed necessary. Conduction velocities were corrected using the formula 2.4 m/s for each degree below 34°C. Electrodiagnostic studies were considered to show evidence for an axonal neuropathy when low-amplitude SNAPs or CMAPs were detected. No studies revealed features of demyelination.
Questionnaire. Since we did not have access to medical records, we relied on information obtained from participants by having each fill out a questionnaire pertaining to medication use, other medical diseases, symptoms associated with weakness, sensory loss, and autonomic function, and history related to the diagnosis of SS based on the European Study Group and American–European Consensus Study Group on Diagnostic Criteria for SS.17,18 For the diagnosis of SS, patients were asked whether they: (1) had dry eyes every day for more than 3 months, had recurrent sensation of sand or gravel in the eyes, or used tear substitute more than three times daily; (2) had dry mouth every day for more than 3 months, had recurrent or persistently swollen salivary glands, or required liquids to aid in the swallowing of dry food; (3) had a positive Schirmer’s or Rose Bengal test; (4) had a salivary gland biopsy showing inflammation; (5) had an abnormal salivary scintigraphy, parotid sialography, or salivary flow study; or (6) had antibodies to Ro(SS-A) or La(SS-B). The diagnosis of SS was determined based on the criteria proposed by the European Study Group18 and the American–European Consensus Group.17 Definite SS is diagnosed when four to six items are positive as long as either item 4 (histopathology) or item 6 (serology) is positive, or if three of six of the objective items (i.e., items 3– 6) are positive. Probable SS is diagnosed if three of six items are positive and the criteria for definite SS are not fulfilled.
Laboratory Evaluation. Blood was drawn and processed for the presence of ANA and SS-A and SS-B antibodies. Enzyme-linked immunoabsorbent assay was also performed to look for antisulfatide, anti– myelin-associated glycoprotein, and antiganglioside antibodies.
Statistics. Fisher’s exact test was used to compare the differences involving the categorical variables between patients with SS and controls. P 0.05 was considered statistically significant.
RESULTS
The mean number of years since diagnosis for patients with SS was 12.5 (range 1– 45). There was no significant difference in age, which was 57 years for subjects and 61 years for controls. However, there was a significant difference in gender; all of the subjects with SS were female compared to only 30% of controls. Immunomodulating drugs were used by 12 of the 22 subjects with SS and included hydroxychloroquine in 11, corticosteroids in 3, and cyclosporine in 3. Other medical conditions reported by patients with SS included osteoarthritis (10), osteopenia (4), hypertension (5), hypothyroidism (3), and anemia (2). Only 1 patient with SS gave a history of diagnosis with neuropathy.
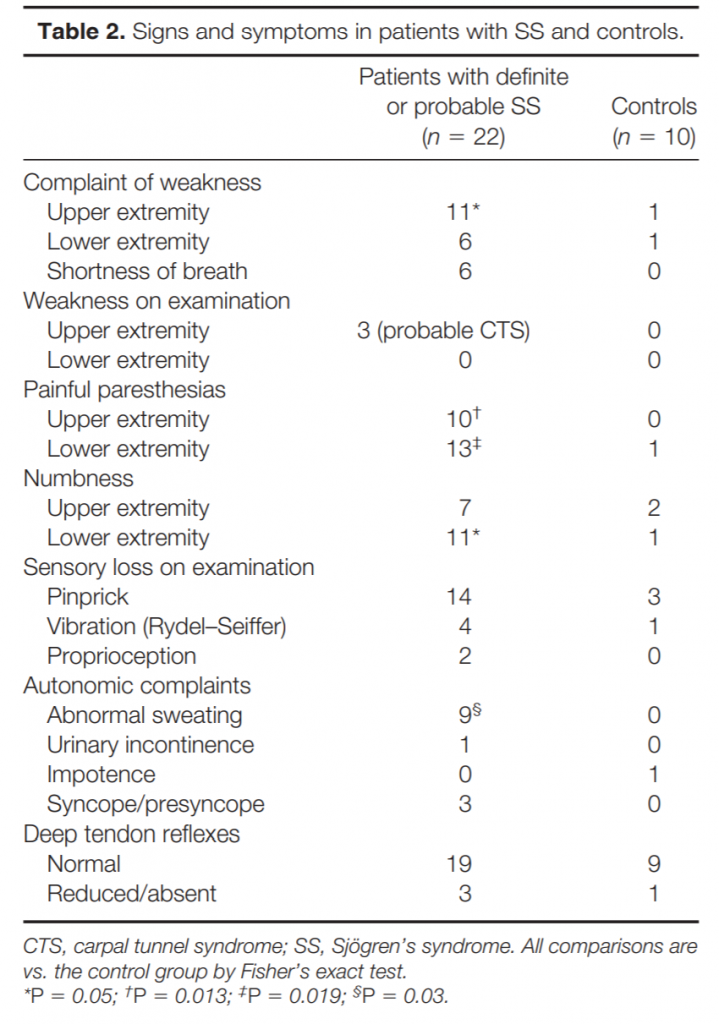
Clinical Characteristics. Although weakness was a common complaint, noted in over 50% of subjects with SS, examination found only mild weakness of thumb muscles in 3 that was thought to be most likely related to carpal tunnel syndrome (Table 2).
Two of these patients also had sensory loss in a median nerve distribution, whereas one had symmetric sensory loss in the hands attributed to a symmetric neuropathy. The complaint of dysphagia, noted in 10 patients with SS, was probably related to the frequent occurrence of dry mouth because neither dysarthria nor facial weakness was noted on examination.
Most differences between SS patients and controls related to small-fiber sensory function (Table 2). Complaints of painful paresthesias were significantly more common in patients with SS than controls, as were complaints of numbness in the feet. As another manifestation of small-fiber dysfunction, sweating complaints, either of hypohidrosis or hyperhidrosis, were also significantly more frequent in patients with SS and were never noted in controls. Distal pinprick loss on examination was twice as common in patients with SS than in controls, although this difference did not reach statistical significance. In contrast, signs of large-fiber sensory function were similar between patients with SS and controls (Table 2). Vibratory or proprioceptive loss on examination was equally common among patients and controls, as were symmetrically reduced DTRs, suggesting that small fibers were preferentially involved.
Electrophysiology. There were no significant differences in electrodiagnostic studies between patients with SS and controls. Abnormal sural SNAP amplitudes (5 V) were noted in 4 patients with SS (18%) and in 2 controls (20%), whereas abnormal peroneal CMAP amplitudes (2 mV) were noted in 5 patients (23%) and 1 control (10%). There were also no significant differences in the mean SNAP or CMAP amplitudes comparing patients with SS and controls. All patients with SS had normal or only borderline abnormal conduction velocities once corrected to a temperature of 34°C. A total of 5 patients with SS (23%) and 2 controls (20%) had NCS and clinical findings thought to be consistent with a sensory or sensorimotor polyneuropathy. In contrast, 10 patients with SS (45%) had symptoms of painful distal paresthesias with isolated small-fiber sensory dysfunction on examination and normal NCSs, whereas no controls had an isolated small-fiber neuropathy (P 0.013). None of the patients with SS had diffusely absent SNAPs with normal CMAPs, consistent with a sensory neuronopathy.
DISCUSSION
Peripheral neuropathy is common in SS, with estimates ranging from 10% to 60% depending on the method of detection, definition of peripheral nervous system disease, and cohort being studied.1– 3,5,6,8,10,11,13,15 Although many different types of neuropathy have been described, most reports list a distal axonal sensory or sensorimotor polyneuropathy as the most common phenotype, accounting for over 50% of cases of peripheral nerve involvement.1–3,5,6,8,10 –13 Manifestations include distal paresthesias and evidence of large-fiber sensory dysfunction on examination and by electrodiagnostic studies. The other type of neuropathy that is commonly reported in SS is a pure sensory neuropathy/ ganglionopathy, thought to be due to large-fiber sensory dysfunction associated with a severe sensory ataxia and at times trigeminal neuropathy.1,5,7,10 –13 In one recent series in which patients were selected from a large tertiary-care neurology department, the diagnosis of sensory ganglionopathy was made in 40%.16
In contrast, in our patients, we found a high prevalence of small-fiber neuropathy, a finding that has only rarely been reported.4,6,9,16 Nearly 50% of our patients with definite or probable SS had complaints of painful distal paresthesias combined with evidence of small-fiber sensory loss with normal large-fiber function as determined by examination and electrodiagnostic studies. In contrast, no control subject had similar abnormalities. Most of the patients with clinical evidence of neuropathy were not diagnosed previously (only 1 patient had a prior diagnosis of neuropathy). This highlights the likely mild or subclinical neuropathy present in many patients with SS3 that may eventually lead to disability especially related to painful distal paresthesias.
Evidence for small-fiber sensory neuropathy was both subjective and objective. Subjective complaints indicating small-fiber involvement in patients with SS included distal painful paresthesias and abnormalities in sweating. Clinical evidence of small-fiber sensory loss included abnormalities on testing for pinprick appreciation in the feet, which were twice as common in patients with SS as in controls. Although the sensory examination is not completely objective, any inherent subjectivity during the examination was controlled for by the fact that our examiners were blinded to the diagnosis of SS. A recent study confirmed that the clinical findings of small fiber sensory loss correlate with the intraepidermal nerve fiber density, an objective measure of small fiber neuropathy.4
Large-fiber sensory dysfunction, as suggested by reduced or absent DTRs and abnormal vibration tested by the Rydel–Seiffer tuning fork and by abnormalities on NCSs, was also common in our group of patients with SS, with evidence of a sensory or sensorimotor polyneuropathy in 5 of 22 (23%). However, this was similar to the incidence of large fiber dysfunction in our control group, 2 of 10 (20%). Only 1 patient in our initial cohort of 29 patients had evidence by examination and on NCSs of isolated diffuse sensory dysfunction consistent with a sensory neuronopathy, although this patient did not fulfill criteria for definite or probable SS.
There are several potential biases in our study design. We cannot be sure that all of the patients who reported a history of SS actually had SS because we were not able to evaluate the primary data. However, all of our patients had a history of SS, over half were treated with immunosuppressive drugs that are typically used in SS, and the majority fulfilled diagnostic criteria for SS. In addition, a majority had laboratory features of SS including antibodies against SS-A or SS-B, or ANA.
The other main drawback to our study relates to our control group, which was small, was not matched for gender with SS patients, and had a relatively high frequency of neuropathy. However, since an overestimation of neuropathy in the control group would tend to lessen any statistical significance when comparing to the patients with SS, we consider our comparisons valid. Other minor drawbacks include the fact that our questionnaire has not been rigorously validated and that limb warming could not be done to help standardize NCSs. However, as both patients with SS and controls were subjected to similar tests, this is unlikely to have caused over- or underrepresentation of neuropathy in either group. The etiology of small-fiber neuropathy in patients with SS is unknown. However, several previously reported patients who initially presented with a small-fiber neuropathy later developed a sensory ataxic neuropathy.16 This suggests that small-fiber neuropathy is on a continuum with large-fiber sensory neuropathy, which at least in some patients has been shown to be due to inflammation within the dorsal root ganglion.12 It is possible that, in some patients with SS and small-fiber neuropathy, inflammation within the dorsal root initially affects those neurons that convey small-fiber modalities. Regardless of the cause, testing different populations of patients with SS will help to determine the true prevalence of small-fiber and other neuropathy subtypes.
Presented in part at the 56th annual meeting of the American Academy of Neurology, April 2004, San Francisco, California.
REFERENCES
- Alexander EL, Provost TT, Stevens MB, Alexander GE. Neurologic complications of primary Sjögren’s syndrome. Medicine 1982;61:247–257.
- Andonopoulos AP, Lagos G, Drosos AA, Moutsopoulos HM. The spectrum of neurological involvement in Sjögren’s syndrome. Br J Rheumatol 1990;29:21–23.
- Barendregt PJ, van den Bent MJ, van Raaij-van den Aarssen VJ, van den Meiracker AH, Vecht CJ, van der Heijde GL, et al. Involvement of the peripheral nervous system in primary Sjögren’s syndrome. Ann Rheum Dis 2001;60:876 – 881.
- Chai J, Herrmann DN, Stanton M, Barbano RL, Logigian EL. Painful small-fiber neuropathy in Sjögren’s syndrome. Neurology 2005;65:925–927.
- Delalande S, de Seze J, Fauchais AL, Hachulla E, Stojkovic T, Ferriby D, et al. Neurologic manifestations in primary Sjögren’s syndrome: a study of 82 patients. Medicine 2004;83: 280 –291.
- Denislic M, Meh D, Popovic M, Kos-Golja M. Small nerve fibre dysfunction in a patient with Sjögren’s syndrome. Neurophysiological and morphological confirmation. Scand J Rheumatol 1995;24:257–259.
- Font J, Ramos-Casals M, de la Red G, Pou A, Casanova A, Garcia-Carrasco M, et al. Pure sensory neuropathy in primary Sjögren’s syndrome. Longterm prospective followup and review of the literature. J Rheumatol 2003;30:1552–1557.
- Gemignani F, Marbini A, Pavesi G, Di Vittorio S, Manganelli P, Cenacchi G, et al. Peripheral neuropathy associated with primary Sjögren’s syndrome. J Neurol Neurosurg Psychiatry 1994;57:983–986.
- Gorson KC, Ropper AH. Positive salivary gland biopsy, Sjögren’s syndrome, and neuropathy: clinical implications. Muscle Nerve 2003;28:553–560.
- Govoni M, Bajocchi G, Rizzo N, Tola MR, Caniatti L, Tugnoli V, et al. Neurological involvement in primary Sjögren’s syndrome: clinical and instrumental evaluation in a cohort of Italian patients. Clin Rheumatol 1999;18:299 –303.
- Grant IA, Hunder GG, Homburger HA, Dyck PJ. Peripheral neuropathy associated with sicca complex. Neurology 1997; 48:855– 862.
- Griffin JW, Cornblath DR, Alexander E, Campbell J, Low PA, Bird S, et al. Ataxic sensory neuropathy and dorsal root ganglionitis associated with Sjögren’s syndrome. Ann Neurol 1990;27:304 –315.
- Lafitte C, Amoura Z, Cacoub P, Pradat-Diehl P, Picq C, Salachas F, et al. Neurological complications of primary Sjögren’s syndrome. J Neurol 2001;248:577–584.
- Martina IS, van Koningsveld R, Schmitz PI, van der Meche FG, van Doorn PA. Measuring vibration threshold with a graduated tuning fork in normal aging and in patients with polyneuropathy. European Inflammatory Neuropathy Cause and Treatment (INCAT) group. J Neurol Neurosurg Psychiatry 1998;65:743–747.
- Mellgren SI, Conn DL, Stevens JC, Dyck PJ. Peripheral neuropathy in primary Sjögren’s syndrome. Neurology 1989;39: 390 –394.
- Mori K, Iijima M, Koike H, Hattori N, Tanaka F, Watanabe H, et al. The wide spectrum of clinical manifestations in Sjögren’s syndrome–associated neuropathy. Brain 2005;128: 2518 –2534.
- Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American–European Consensus Group. Ann Rheum Dis 2002;61:554 –558.
- Vitali C, Bombardieri S, Moutsopoulos HM, Balestrieri G, Bencivelli W, Bernstein RM, et al. Preliminary criteria for the classification of Sjögren’s syndrome. Results of a prospective concerted action supported by the European Community. Arthritis Rheum 1993;36:340 –347.